2024年最值得关注的13款药物(附销量预测)
近日,数据分析公司科睿唯安(Clarivate)发布《2024年最值得关注的药物预测》,列出了13种于近期获批或即将获批的、具有改变医药市场的潜力药物,种类涵盖小分子、单抗、双抗、ADC、融合蛋白、基因(编辑)疗法、疫苗;这些药物有望在未来五年成为重磅炸弹和/或改变现有的治疗模式。
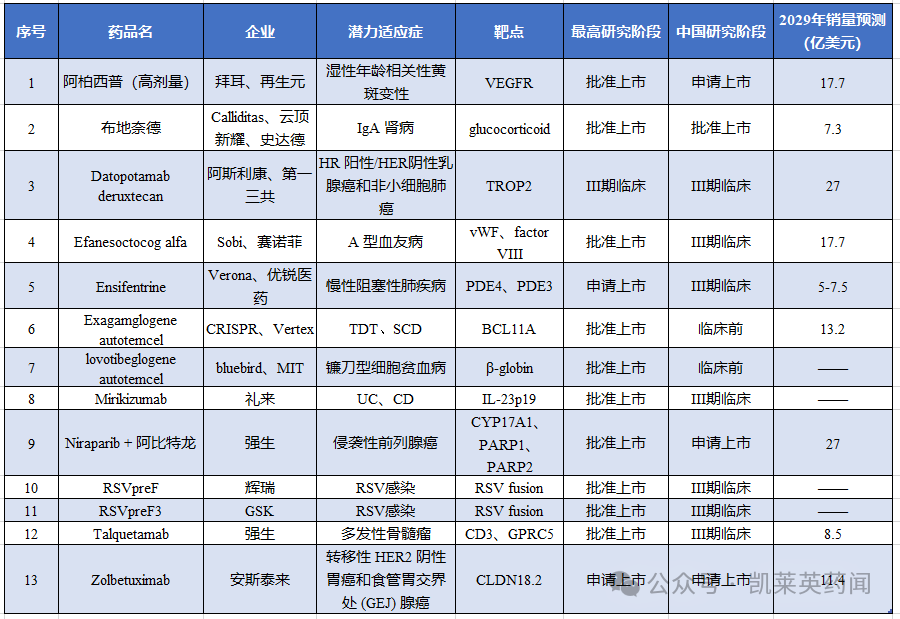
2024年最值得关注的13款药物
1、拜耳/再生元:Eylea HD
作用机制:抗VEGF药物
适应症:年龄相关性黄斑变性(wADM)
阿柏西普是一种全人类的重组融合蛋白,由VEGF受体1的第二免疫球蛋白(Ig)结合域和VEGF受体2的第三Ig结合域组成,与人类IgG1的Fc区融合。与其他的抗血管内皮生长因子疗法不同,它除了和所有VEGF-A异构体、VEGF-B,还能够与胎盘生长因子(PlGF)结合。除此之外,阿柏西普的另一个特点是,它对VEGF显示出非常高的亲和力,亲和力(Kd)达到0.5pM,强于雷珠单抗、贝伐单抗以及自身VEGF受体。这意味着阿柏西普即使在低浓度下也能有效阻断VEGF,因此有着更长的作用时间,从而可以延长给药间隔。
自2011年获批用于wADM的治疗后,阿柏西普其他适应症被相继开发出来。2014年7月,阿柏西普注射液获FDA批准用于治疗糖尿病性黄斑水肿;10月,获FDA批准用于治疗视网膜静脉阻塞(RVO)后的黄斑水肿;2015年3月,获批用于糖尿病黄斑水肿患者的糖尿病视网膜病变;2018年,FDA批准阿柏西普新注射剂型用于治疗湿性年龄相关性黄斑变性;2019年3月,获批用于糖尿病视网膜病变。除了眼科适应症, 2012年,FDA批准阿柏西普(ZaltrapÒ)用于治疗转移性结直肠癌,与氟尿嘧啶、亮丙瑞林和伊立替康(Onivyde或Camptosar)联合使用等等……
阿柏西普类抗血管生成药物(例如雷珠单抗)存在的最大问题是因为需要在玻璃体内给药且需要频繁注射和检测产生的患者依从性问题;罗氏Vabysmo(faricimab)则将时间从原来的八周延长到了4个月,是阿柏西普的两倍,存在着明显的优势。8 mg剂量的阿柏西普则是缩小阿柏西普和Vabysmo之间给药间隔差距的方法之一。
阿柏西普高剂量(Eylea HD)最早于2023年8月获批,已陆续被美国、欧盟、日本授予上市许可。研究显示,阿柏西普在高剂量8mg长间隔Q12W/Q16W治疗DME和wAMD两个试验中,都与阿柏西普(低剂量短间隔组)展现出非劣效的视觉改善和相当的安全性。
科睿唯安推测,2029 年该产品在湿性AMD 患者中的预期销售额将达到17.7亿美元。
2、Calliditas/云顶新耀:布地奈德迟释胶囊
作用机制:糖皮质激素
适应症:IgA 肾病
布地奈德是一种糖皮质激素,具有强糖皮质激素活性和弱盐皮质激素活性,首过代谢程度极高;通过覆以肠溶包衣,使Nefecon可以完整无损地到达回肠,靶向作用于回肠末端的黏膜 B 细胞(包括派尔集合淋巴结),从而减少诱发 IgA 肾病的半乳糖缺陷的 IgA1 抗体( Gd-IgA1 )产生。该药物的商品名为Nefecon,是全球首个IgA 肾病的靶向治疗药物,用于具有进展风险的成人原发性 IgA 肾病,降低蛋白尿水平。该药物已于2021年获FDA加速批准上市,于2022年获EMA附条件上市许可批准,于2023年11月获NMPA批准上市,用于治疗具有进展风险的原发性IgA肾病成人患者。
Nefecon是由Calliditas开发,2019年6月,云顶新耀与Calliditas签订独家授权许可协议,获得在大中华地区和新加坡开发以及商业化 Nefecon的权利;该协议于 2022 年 3 月扩展,将韩国纳入云顶新耀的授权许可范围。
该药物在国内获批主要基于一项3期、随机、双盲、安慰剂对照、多中心NeflgArd研究结果,该研究旨在两年研究期间(9个月Nefecon或安慰剂治疗+15个月停药随访)评估 Nefecon,或与RAS 抑制剂联用的疗效和安全性。根据2023年11月在美国肾脏病学会肾脏周会上的披露的3期中国亚组数据,结果显示:第24个月时, eGFR较基线的平均绝对变化表明,中国患者中耐赋康相较于安慰剂可以延缓肾功能衰退达66%(VS. 全球50%),其中:(1)第24个月时,中国人群安慰剂组eGFR较基线平均下降了20.97 mL/min/1.73 m2,全球数据下降12 mL/min/1.73 m2;中国人群安慰剂平均UPCR自基线上升了18.6%,而全球人群为下降1%。此外,在中国安慰剂组中更多的患者有镜下血尿。
目前IgA 肾病的治疗方案仍以对症治疗为主,包括血管紧张素转换酶抑制剂和血管紧张素受体阻滞剂等,激素等则被建议用于改善远期预后,治疗方案整体疗效有限且副作用较大,患者有较大的新疗法需求。基于Nefecon的吸收特性,即使长期使用,其所产生的副作用可能仍然比其他皮质类固醇更少。据科睿唯安推测,2029 年该产品的预期销售额将达到7.3亿美元。
3、AZ/第一三共:Datopotamab deruxtecan
作用机制:靶向Trop2的ADC
适应症:肺癌
Datopotamab deruxtecan(Dato-DXd,DS-1062a)是一种靶向Trop2的ADC药物,是人源化抗TROP2 IgG1单克隆抗体与有效载荷强效拓扑异构酶I抑制剂通过稳定的四肽可裂解连接子偶联而成,药物/抗体比率(DAR)值为4。
TROP2是一种跨膜糖蛋白,广泛表达于多种实体瘤中,包括腺癌(64%)和鳞状细胞癌(75%)等;目前尚无TROP2靶向ADC获批用于治疗肺癌患者。除此以外,三阴性乳腺癌(TNBC)被认为是最具侵袭性的乳腺癌亚型,大约15%的乳腺癌被认为是三阴性,与其他乳腺癌亚型相比,疾病复发率更高且预后更差;TROP-2在三阴性乳腺癌患者中表达超80%,明显高于在其他疾病类型,成为乳腺癌治疗的明星靶点。
据不完全统计,目前Dato-DXd针对肺癌的TROPION-Lung01 III 期试验数据,以及针对乳腺癌的TROPION-Breast01 III 期试验以及BEGONIA Ib/II 期试验数据,均展示出良好的治疗潜力。据科睿唯安推测,2029 年该产品在乳腺癌和非小细胞肺癌的预期销售额将达到27亿美元。
4、Sobi/赛诺菲:Efanesoctocog alfa
作用机制:融合蛋白
适应症:A 型血友病
Efanesoctocog alfa是一种新型融合蛋白,基于创新的Fc融合技术开发,通过添加vWF的一个区域和XTEN多肽来延长其在血液循环中的时间。Efanesoctocog alfa具有独特的结构,能够独立于vWF而稳定发挥作用,且半衰期延长至约47h,在一周一次的预防性治疗方案中,为一周的大部分时间提供接近正常水平的因子活性。
相对于目前的VIII因子模拟疗法和仍在临床试验中的止血平衡疗法,efanesoctocog alfa的主要优势是作为VIII因子的替代品用于治疗血友病患者的急性出血,并可通过标准实验室化验,以在必要时进行监测和剂量调整。Efanesoctocog alfa是目前唯一一种已被证明突破血管性血友病因子上限的疗法。
2023年2月,efanesoctocog alfa(商品名:Altuviiio)获FDA批准上市,该获批主要基于一项开放标签、非随机干预研究,评估每周一次Altuviiio,在 12 岁或以上 (n=159) 的严重血友病 A 患者中的安全性、有效性和药代动力学,这些患者既往接受过因子 VIII 替代疗法治疗。主要疗效终点是平均年化出血率(ABR),关键的次要终点是比较Altuviiio每周预防治疗和使用其它VIII因子预防治疗的ABR。
结果显示:(1)每周一次的Altuviiio预防达到主要终点,为重度血友病A患者提供显著的出血保护,平均年出血率(ABR)为0.70,中位ABR为0.0。此外,研究达到了关键的次要终点,与先前的预防治疗相比,ABR显著降低了77%。(2)其他数据显示,Altuviiio能够预防关节出血,中位年化关节出血率为0。在一周的大部分时间里,提供的平均因子VIII活性大于40%,在第7天大于10%;这些水平与低出血风险有关。(3)Altuviiio耐受性良好,最常见的副作用(>10%)是头痛和关节痛。
此外,在既往接受过治疗的12岁以下严重血友病A患者中每周一次Altuviiio的安全性,有效性和药代动力学XTEND-Kids研究显示:(1)研究在74名参与研究的儿童中未检测到因子VIII抑制物,其中65名儿童至少接触了50天。(2)Efanesoctocog alfa在整个每周给药间隔期间,提供高持续的因子VIII水平,年化出血率(ABR)中位数为0.00(四分位数范围:0.00-1.02),估计平均ABR(95%置信区间)为0.89(0.56-1.42)。
据科睿唯安推测,2029 年该产品的预期销售额将达到17.7亿美元。
5、Verona/优锐:Ensifentrine
作用机制:PDE3/PDE4抑制剂
适应症:慢性阻塞性肺疾病
Ensifentrine(恩塞芬汀)是一款磷酸二酯酶蛋白吸入型双靶点PDE 3/4抑制剂,双重抑制机理使其能够凭借单个化合物同时实现支气管扩张和抗炎效应。中性粒细胞炎症是慢性阻塞性肺病(COPD)的最主要作用途径,通常对类固醇治疗应答不理想。Ensifentrine 是一种对中性粒细胞也非常有效的药物,因此这也是该药物能够减少 COPD 主要炎症成分的潜在重要途径。
2021年,优锐医药与Verona Pharma签订协议,获得恩塞芬汀在大中华区域开发与商业化的独家权益。2023年4月,优锐医药宣布ENHANCE–CHINA中国三期临床试验完成首例患者给药。
2023年9月,FDA已接受审评恩塞芬汀的新药上市申请,用于维持治疗COPD患者;并设定PDUFA日期为2024年6月。如果获得批准,恩塞芬汀有望成为COPD领域10多年来批准的全新机制药物。该新药上市申请包含约3,000名受试者在ENHANCE-1与ENHANCE-2三期临床研究取得的积极试验数据;两项研究的结果显示,恩塞芬汀组患者的平均1秒平均用力呼气量(FEV1)分别增加了87mL和94mL,COPD恶化率分别降低了36%和43%;两项研究展现出对肺部功能指标的有效改善,降低COPD的病情恶化率。
作为所有新型药物(包括生物制品)中处于最快开发阶段的药物,恩塞芬汀最有可能被用作长效支气管扩张疗法的附加治疗。据科睿唯安推测,2029 年该产品的预期销售额将达到5-7.5亿美元。
6、CRISPR/Vertex:Exagamglogene autotemcel
作用机制:基因编辑疗法
适应症:β地中海贫血和镰状细胞性贫血
Exagamglogene autotemcel(exa-cel,Casgevy)是CRISPR与Vertex 联合研发的体外基因编辑疗法,开发适应症为镰状细胞病 (SCD)和 β-地中海贫血症(TDT)。通过体外基因编辑,研发团队对CD34+ HSPC 细胞中红细胞(erythroid)特异的BCL11A 基因的加强子(enhancer)进行抑制,以此得到改造后的细胞并给患者进行回输。BCL11A蛋白是一种调控因子,通常在出生后抑制胎儿血红蛋白的产生,因此通过BCL11A基因的编辑减少其蛋白产物的合成,可以促进造血干细胞产生更多携带胎儿血红蛋白的红细胞,以逐步取代成人的镰状红细胞。
Exa-cel 针对SCD和TDT分别开展了临床研究,结果显示:在针对TDT 适应症的研究中,受试者接受exa-cel 治疗后,42/44 患者已停止输注红细胞。其中持续时间最长的已有36.2 个月没有进行输注;2 位还未停止输注的患者,所需输注的红细胞量已明显减少,分别下降了75%、89%。在针对SCD 适应症的研究中,接受exa-cel 治疗后,所有31 个患者均未出现血管闭塞危象VOC,其中持续时间最长的已有32.3 个月未出现VOC。
此外,Exa-cel 治疗后,含HbF的细胞比例明显提升,TDT 组由基线的约10%提升至90%-100%区间,SCD组由基线的约20%提升至90%-100%。且提升后比例稳定维持在高位区间,TDT 组最长观察期已达36 个月,SCD组30 个月。
在安全性上,与白消安清髓术和自体造血干细胞移植(HSCT)相似。许多不良事件可能与配套的化疗(白消安)相关。
该药物是全世界首款获批上市的CRISPR基因编辑疗法,据科睿唯安推测,2029 年该产品的预期销售额将达到13.2亿美元。
7、bluebird/MIT:lovotibeglogene autotemcel
作用机制:基因疗法
适应症:镰刀型细胞贫血病
Lovotibeglogene autotemcel(lovo-cel,Lyfgenia)的工作原理是通过慢病毒载体将β珠蛋白基因的功能拷贝递送至患者的血液干细胞中,以产生常规的红血球蛋白;该药物通过将改良形式的β-珠蛋白基因(βA-T87Q珠蛋白基因)的功能,拷贝添加至患者自身的造血干细胞(HSCs)中,使患者的红细胞产生抗镰状血红蛋白(HbAT87Q),从而降低HbS的比例。
此前,FDA已授予lovo-cel孤儿药资格、快速通道资格、再生医学先进疗法认定(RMAT)和罕见儿科疾病认定。2023年12月,FDA批准lovo-cel的生物制品许可申请(BLA),用于治疗12岁及以上患有镰刀型细胞贫血病(SCD)且有血管闭塞事件(VOE)史的患者。该BLA主要是基于I/II期HGB-206研究C组(n=36)的疗效数据。结果显示,32例患者接受输注后在可评估的6-18个月内严重血管闭塞发作(VOEs)完全消退,30/32例患者(94%)的严重血管闭塞事件得到解决,28/32例患者(88.2%)血管闭塞事件完全消退。在安全性上,最常见的≥3级不良反应(发生率≥20%)为口腔炎、血小板减少、中性粒细胞减少、发热性中性粒细胞减少以及贫血等。
在此之前,镰状细胞病可用的治疗方法很少,唯一的治疗方法是骨髓移植;蓝鸟生物将该产品定价为310万美元。
8、礼来:Mirikizumab
作用机制:IL-23单抗
适应症:溃疡性结肠炎、克罗恩病
Mirikizumab 是一种白细胞介素 23(IL-23)p19 拮抗剂,可与IL-23的p19亚基结合,进而阻断IL-23介导的炎症反应。2023年10月,该药物获FDA批准用于治疗成人中度至重度活动性溃疡性结肠炎(UC);同月,公司宣布,该药物用于治疗患有中度至重度活动性克罗恩病(CD)的成人的VIVID-1研究中达到了共同主要终点和所有主要次要终点,公司计划在 2024 年向FDA提交mirikizumab用于治疗CD的上市申请。
两项 III期随机试验(LUCENT-1和LUCENT-2)验证了在UC人群中的试验结果,显示与安慰剂相比,mirikizumab在 UC 患者的诱导和维持治疗后的临床缓解方面更有效。
其中LUCENT-1 纳入的1,281 名患者,按照3:1 的比例随机接受 300 mg 静脉注射mirikizumab 或安慰剂,每 4 周接受一次,持续12 周。在诱导试验中没有反应的患者允许在维持试验的前 12 周内接受开放标签 mirikizumab 作为延长诱导。结果显示:在 LUCENT-1诱导试验的 1,200 多名患者中,接受mirikizumab 治疗的患者中有 24.2% 在第 12周出现临床缓解,而安慰剂组的仅为 13.3%(P<0.001)。
LUCENT-2 纳入了 544 名在 LUCENT-1 中对 mirikizumab 有反应的患者,并以 2:1 的比例随机分配,每 4 周接受200 mg mirikizumab 或安慰剂,持续 40 周。维持临床缓解的定义在诱导试验中使用mirikizumab 获得的持续临床缓解。结果显示:分别有 49.9% 和 25.1% 在第 40 周出现临床缓解(P<0.001),并且维持临床缓解的比例超过60%。次要终点,包括临床反应、内镜缓解、组织学指标和排便紧迫感的改善也达标。
除此以外,VIVID-1研究验证了mirikizumab用于治疗患有中度至重度活动性CD的成人的疗效;在该研究中,所有接受药物治疗的患者在12周后继续按原方案接受治疗,直至研究的第52周结束。而在第12周未获得疗效的安慰剂组患者(无应答者)则转为接受盲法mirikizumab的治疗。试验结果显示,与接受安慰剂的患者比较,接受mirikizumab治疗的患者在第12周和第52周获得临床缓解的比例显著更高。
Mirikizumab是首款治疗UC的 IL-23p19 单抗,亦可能成为获批用于治疗CD的第三种药物;此前获批UC的两款IL-23单抗分别是乌司奴单抗和利生奇珠单抗。分析师预测,Mirikizumab将在2028年达到12亿美元的年销售量。
9、强生:Niraparib +阿比特龙
作用机制:CYP17A1/ PARP抑制剂
适应症:侵袭性前列腺癌
尼拉帕利(niraparib,Zejula)是一种PARP抑制剂,通过抑制PARP酶的活性,使PARP-DNA复合物的形成增加,导致DNA损伤、凋亡和细胞死亡;阿比特龙(abiraterone acetate,Zytiga)是一种CYP17抑制剂,常联合泼尼松用于既往接受多西他赛治疗的转移性去势抵抗性前列腺癌患者。2023年4月,强生旗下杨森制药宣布Akeega获EMA上市许可,这是该药物在全球范围内的首次获批;Akeega是由尼拉帕利和醋酸阿比特龙组合而成的复方片剂。
MAGNITUDE研究是一项随机双盲研究,纳入423例符合条件的HRR基因突变患者,其中212例接受尼拉帕利治疗,211例接受安慰剂治疗。治疗包括每日200 mg尼拉帕利+每日1 g标准阿比特龙+每日10 mg泼尼松,或每日200 mg安慰剂+每日1 g标准阿比特龙+每日10 mg泼尼松。研究的主要终点是rPFS(影像学无进展生存期)。
结果显示,在BRCA突变亚组中,尼拉帕利和安慰剂的rPFS分别为16.6个月和10.9个月,有显著的统计学改善(HR 0.53;95%CI 0.36,0.79;p=0.0014)。在BRCA突变亚组的探索性分析中,尼拉帕利和安慰剂的OS分别为30.4个月和28.6个月。
在mCRPC患者中,通常使用雄激素剥夺疗法来阻断男性性激素的作用,但前列腺癌仍会生长并扩散到身体的其他部位。大约10-20%的晚期前列腺癌男性会在五年内发展为去势抵抗性前列腺癌(CRPC),其中至少有84%的男性在CRPC诊断时会有转移。在CRPC诊断时没有转移的患者中,33%可能在两年内发生转移。大约10%-15%的mCRPC患者携带BRCA1/2基因突变。据科睿唯安推测,2029 年上述复方制剂的预期销售额将达到27亿美元。
10、辉瑞:RSVpreF
作用机制:重组蛋白疫苗
适应症:RSV感染
RSVpreF(Abrysvo)属于二价呼吸道合胞病毒(RSV)疫苗,通过注射怀孕32 至36 周妇女来实现对出生至6 个月婴儿的RSV 预防。2023 年5 月,辉瑞宣布FDA已批准Abrysvo(PF-06928316)上市,可用于预防60 岁及以上人群出现由RSV 引起的下呼吸道疾病。2023 年8月,该药物的新适应症获批,通过孕妇主动免疫,预防出生至6 个月大的婴儿患上RSV 相关下呼吸道疾病(LRTD),这是FDA 批准的首款保护婴儿在出生至6 个月内免受RSV引起的LRTD 和严重LRTD 的孕产妇接种疫苗。
在针对老年群体的3期临床试验中,辉瑞共招募了3.4万多名60岁及以上的老年人,以测试该疫苗的安全性和有效性。结果显示,与安慰剂相比,辉瑞的RSVpreF疫苗在预防至少有两种症状的RSV相关下呼吸道疾病方面的有效率为66.7%(96.66% CI:28.8-85.8),在预防有三种或以上症状感染方面的有效率为85.7%(96.66% CI:32.0-98.7)。对于避免发生病毒相关的急性呼吸道疾病有效率为62.1%(95% CI:37.1-77.9)。
针对婴儿的MATISSE 临床3 期试验数据显示,研究约近7400 名孕妇入组,这些孕妇在妊娠24-36 周时接受RSVpreF 疫苗或安慰剂单次肌肉注射。在婴儿出生后的前90 天内,RSVpreF 对LRTD的保护效力为57.1%,对严重LRTD 的保护效力达81.8%;在婴儿出生后的前180 天内,RSVpreF 对下LRTD的保护效力为51.3%,对严重LRTD 的保护效力达69.4%。
上市后首个季度,辉瑞实现3.75亿美元的销售额,实现疫苗大单品的快速放量。
11、葛兰素史克:RSVpreF3
作用机制:重组蛋白疫苗
适应症:RSV感染
RSVpreF3(Arexvy)由RSV融合前(prefusion)F 糖蛋白(RSVPreF3)与GSK 专有的佐剂AS01E组合而成,2023 年5 月,FDA批准Arexvy用于预防60 岁及以上人群因感染RSV 引发的下呼吸道疾病(LRTD),是全球首款突破性获批上市的用于老年人RSV疫苗。
FDA此次批准是基于GSK具有里程碑意义的AReSVi 006(成人呼吸道合胞病毒)III期试验数据研究。该研究是一项全球性、多中心、随机、双盲、安慰剂对照的临床试验,共纳入24966例60岁及以上老年人受试者,旨在评估单剂量和每年接种Arexvy(N=12467)对比安慰剂(N=12499)对RSV-LRTD的预防效果。研究的主要终点为第一个RSV流行季期间单剂量Arexvy对RSV-LRTD的预防效力。
结果显示,Arexvy的总体疫苗效力为82.6%(96.95% CI:57.9–94.1),具有统计学意义和临床意义,达到了临床主要终点。对严重RSV-LRTD(定义为至少2种症状)的预防效力为94.1%(95% CI:62.4–99.9);对RSV-A亚型和RSV-B亚型的预防效力一致(84.6% vs 80.9%)。在有并发症的老年人群体(N=9798)中,Arexvy的预防效力为94.6%(95% CI:65.9–99.9);在70-79岁老年人群体(N=8974)中,疫苗的预防效力为93.8%(95% CI:60.2-99.9)。安全性方面,Arexvy具有良好的耐受性和安全性。研究中报告的不良事件(AE)通常是轻度至中度且短暂的,最常见的AE是注射部位疼痛(60.9%)、疲劳(33.6%)、肌痛(28.9%)和头痛(27.2%)。
上市后首个季度,GSK实现7.1 亿英镑的销售额,销量超过辉瑞。据科睿唯安推测,未来五年 RSV 疫苗和预防治疗的潜在市场价值为 100 亿美元。
12、强生:Talquetamab
作用机制:靶向PRC5D /CD3双抗
适应症:多发性骨髓瘤
Talquetamab是一款在研的first-in-class现货型双特异性T细胞结合抗体,能同时靶向多发性骨髓瘤(MM)细胞上的GPRC5D和T细胞上的CD3。GPRC5D名为G蛋白偶联孤儿受体,其在恶性浆细胞上过表达,在正常组织的表达仅限于皮肤和睾丸,且其表达水平与BCMA靶点相对独立。该药物通过激活CD3阳性T细胞,诱导T细胞对GPRC5D阳性MM细胞进行杀伤。
2023年8月,Talquetamab获美国FDA批准上市,用于先前至少接受过4种治疗(包括蛋白酶体抑制剂、免疫调节剂和CD38抗体)的复发或难治性多发性骨髓瘤(MM)成人患者,成为全球范围内首个获批上市的靶向GPRC5D的抗体产品。该获批是基于2期临床试验MonumenTAL-1,其中187例既往至少接受过四线治疗但未接受过T细胞定向治疗的复发/难治性MM患者接受不同剂量的Talquetamab治疗后,均收获了有意义的疾病总缓解率。
其中,每2周接受1次Talquetamab(0.8mg/kg)皮下注射患者的客观缓解率(ORR)为73.6%,约85%的患者治疗应答时间在9个月及以上。中位随访时间近6个月时,有58%的患者收获了非常好的部分缓解(VGPR)或更优疗效,其中33%的患者收获了疾病的完全缓解(CR)或更优疗效。此外,每周接受1次Talquetamab(0.4mg/kg)皮下注射患者的ORR达73.0%,中位缓解持续时间(mDOR)9.4个月。从患者首次出现临床缓解开始,中位随访时间近11个月时,57%的患者收获了VGPR或更优疗效,其中35%的患者收获了CR或更优疗效。
高治疗率、针对 R/R MM的潜在多线治疗,以及某些治疗方案的治疗持续时间过长推动了这个市场的大规模扩张;据科睿唯安推测,2029 年该药物的预期销售额将达到8.5亿美元。
13、安斯泰来:Zolbetuximab
作用机制:靶向CLDN18.2单抗
适应症:胃癌或胃食管交界处腺癌
Zolbetuximab是一种在研的靶向Claudin 18.2 (CLDN18.2)的首创嵌合IgG1单克隆抗体(mAb),可与跨膜蛋白CLDN18.2结合。临床前研究表明,这种结合作用通过激活两种不同的免疫系统途径--抗体依赖性细胞毒性(ADCC)和补体依赖性细胞毒性(CDC)诱导癌细胞死亡。
2023年7月,安斯泰来宣布FDA已接受并授予zolbetuximab 的生物制剂许可申请 (BLA) 优先审查,用于一线治疗局部晚期不可切除或转移性 HER2 阴性胃癌或胃食管交界处(GEJ)腺癌,PDUFA的目标行动日期为2024年 1 月 12 日。本次BLA 是基于 3 期SPOTLIGHT 和 GLOW 临床试验的结果。
3期SPOTLIGHT评估了zolbetuximab与mFOLFOX6(一种包括奥沙利铂、亚叶酸钙和氟尿嘧啶的药物联合方案)联用的有效性和安全性。研究显示达到了其主要终点,即与安慰剂+mFOLFOX6相比,zolbetuximab+mFOLFOX6治疗的患者的无进展生存期(PFS)具有统计学意义;此外,该研究还满足了次要终点,即与安慰剂+ mFOLFOX6相比,zolbetuximab + mFOLFOX6治疗的患者的总生存期(OS)具有统计学意义。在zolbetuximab联合mFOLFOX6治疗的患者中,最常见的治疗紧急不良事件是恶心、呕吐和食欲减退。
3期GLOW 旨在评估zolbetuximab联合CAPOX(一种包括卡培他滨和奥沙利铂联合化疗用药方案)一线治疗CLDN18.2阳性、HER2阴性的局部晚期不可切除或转移性胃癌或GEJ腺癌患者对比安慰剂联合CAPOX的安全性和有效性。结果显示:(1)达到了主要终点:zolbetuximab与CAPOX的联合用药组将疾病进展或死亡风险降低了31.3%;中位无进展生存期方面,治疗组和安慰剂组分别为8.21个月和6.80个月。(2)达到了关键次要终点:zolbetuximab与CAPOX联合用药组显著延长了总生存期(OS),将死亡风险降低了22.9%(风险比=0.771;95%CI:0.615-0.965;P值=0.0118)。中位总生存期方面,治疗组和安慰剂组分别为14.39个月和12.16个月。(3)安全性方面:两组严重不良事件(TEAE)的发生率相近,与以往研究一致,zolbetuximab治疗组与安慰剂组分别为47.2%对49.8%。
转移性 HER2 阴性胃癌和GEJ腺癌对新的有效治疗方法的巨大需求明显未得到满足。zolbetuximab 有望成为进入肿瘤市场的首个 CLDN18.2 抑制剂,为转移性HER2阴性胃或 GEJ 腺癌的一线治疗提供急需的生物标志物驱动选择。据科睿唯安推测,2029 年该药物的预期销售额将达到11.4亿美元。
参考资料
1、科睿唯安、各公司官网
2、佰傲谷BioValley、博药、药品咨询、医药地理、新药之光
3、民生证券
本文出处:https://jk.yebaike.cn/view/70742.html
 微信扫一扫
微信扫一扫