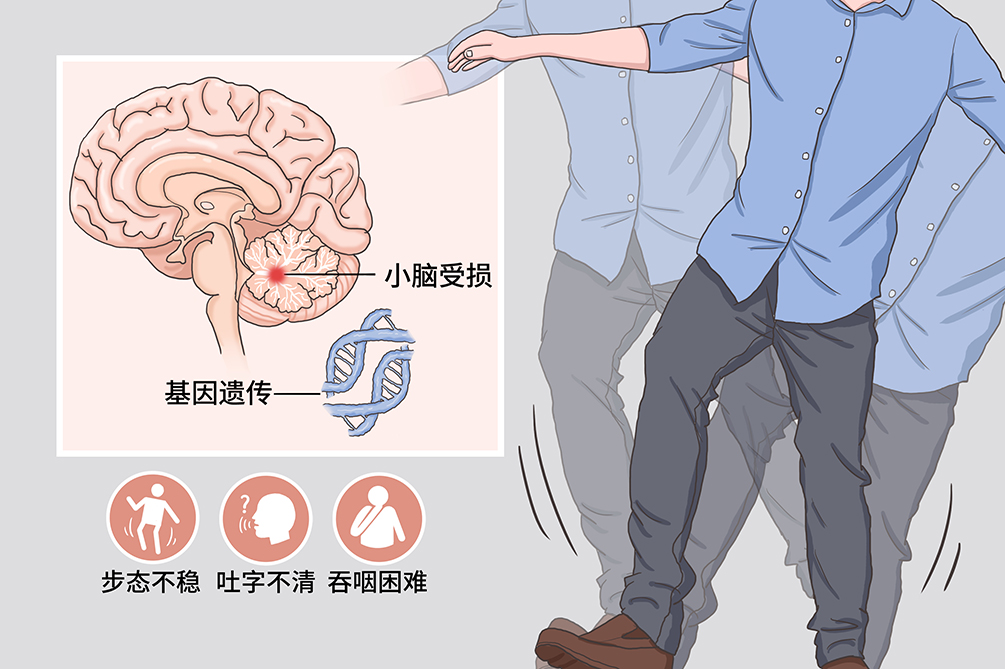
遗传性共济失调
遗传性共济失调是常染色体异常引起的共济运动障碍为突出表现的中枢神经系统变性疾病。本病进展缓慢、有家族遗传史。本病较罕见,临床表现为平衡障碍、进行性肢体协调运动障碍、步态不稳、构音障碍、眼球运动障碍等,并可伴有复杂的神经系统损害,如锥体系、锥体外系、视觉、听觉、脊髓、周围神经损害,亦可伴大脑皮质功能损害,如认知功能障碍和(或)精神行为异常等。该病因尚不明确,大都有遗传史、家族史,具有家族系谱。目前治疗主要采用对症和支持治疗,本病的死亡率和致残率较高。
根据共济失调类型的不同分为三类
脊髓型
Friedreich型共济失调:常染色体隐性遗传,青少年起病,初始行走不稳,渐出现后索损害的症状,Romberg征(+),睁眼可以改善。继之脊髓小脑束受累,出现步基宽、蹒跚步态、定向性震颤和小脑性构音障碍。肢体肌张力降低,腱反射减低或消失,下肢沉重。部分病人可伴有弓形足、脊柱侧弯及其他畸形,个别病人可有心脏异常。
遗传性痉挛性截瘫:常染色体显性或隐性遗传或性连遗传。儿童期起病,男性多见,主要为锥体束受损,多为下肢呈缓慢加重的痉挛性瘫、剪刀状步态。无感觉障碍,上肢很少受累,可伴有原发性视神经萎缩或视网膜色素变性。
小脑型
Marie型共济失调:常染色显性遗传,成年起病,自下肢开始出现小脑型共济失调而无感觉障碍,言语常顿挫或暴发,可有锥体束征及欣快,智力减退。
橄榄小脑桥脑萎缩:常染色体显性遗传,中年后起病,除小脑型共济失调和构音障碍外有早期尿失禁,部分病人有智能减退和锥体外系症状,如帕金森综合征等,但无眼球震颤。
脊髓小脑型
主要有共济失调-毛细血管扩张症。常染色体隐性遗传,婴儿期发病,小脑型共济失调,构音障碍,皮肤、颜面毛细血管扩张,多数伴有舞蹈样手足徐动,随年龄增长而明显。青春期后出现深感觉消失等脊髓后索症状,和病理反射阳性。可因免疫缺陷而反复发生呼吸道感染,晚期有肺部广泛纤维化、肺功能不全等。
按遗传类型分类可分为以下2种
常染色体显性遗传
橄榄-脑桥-小脑萎缩Ⅰ、Ⅲ、Ⅳ、Ⅴ型。
Machado-Joseph’s disease(MJD)。
小脑实质变性Ⅰ型和Ⅳ型。
脊髓脑桥变性。
遗传性痉挛性截瘫。
发作性共济失调。
后索性共济失调。
常染色体隐性遗传
橄榄-脑桥-小脑萎缩Ⅱ、Ⅵ型。
小脑实质性变性Ⅱ、Ⅲ、Ⅳ型。
肌阵挛性小脑性共济失调,或称Ramsay-Hunt综合征。
共济失调性毛细血管扩张症。
Friedreich型共济失调。
病因目前该病病因和发病机制尚未阐明,认为可能主要与以下遗传因素有关,如酶缺乏、缺陷,生化缺陷、线粒体功能缺陷、DNA修复缺陷。
主要病因酶缺乏、缺陷
多见于丙酮酸脱氢酶复合体、氨基己糖苷酯酶A、鸟氨酸氨甲酰基转移酶等缺乏。
生化缺陷
如共济失调伴选择性维生素E缺乏症中的维生素E缺乏,如β-脂蛋白缺乏症中的β-脂蛋白缺乏。血棘红细胞增多和血VE低,如Hartnup,病中的氨基酸尿症。
三核甘酸动态突变
突变形式可表现为突变无义、缺失突变。动态突变是一种新型的突变形式,其是由于DNA中的基重复单位拷贝数不稳定扩增而导致。正常情况下,重复单位拷贝数有一定限制,而在动态突变下,则大大增加。
线粒体功能缺陷
线粒体是给细胞提供能量的细胞区器。线粒体病是一组由MTDNA或核DNA(NDNA)缺陷导致MT结构和功能障碍,ATP合成不足所致的多系统疾病。线粒体病是由核或基因缺陷引起。
DNA修复缺陷
与DNA修复有关的共济失调有AT、着色性干皮病(xP)、Cockayne综合征。
流行病学据统计,遗传性共济失调本病约占整个神经遗传病的10%。大多数在20~40岁发病,婴幼儿和老年人发病较少。其遗传方式以常染色体显性遗传方式为最多。
好发人群该病好发于20~40岁人群。
症状该病主要表现为共济运动障碍,伴有复杂的神经系统损害。神经系统表现包括平衡障碍、进行性肢体协调运动障碍、步态不稳、构音障碍、眼球运动障碍等,并可伴有复杂的神经系统损害,如锥体系、锥体外系、视觉、听觉、脊髓、周围神经损害,亦可伴大脑皮质功能损害,如认知功能障碍和(或)精神行为异常等。还可以出现神经系统以外的病变,如心脏病变、内分泌代谢异常、骨骼畸形、皮肤病变等。
典型症状步态不稳
是最常见的首发症状,出现醉酒样或剪刀步伐。
吐字不清
构音障碍为遗传性共济失调的特征之一,患者主要表现为发音生硬(爆发性言语)、缓慢,单调而含糊,构音不清、音量强弱不等,或时断时续,呈吟诗样语言。病情进展至晚期时,几乎所有患者均出现运动失调性构音障碍。
吞咽困难和饮水呛咳
是由于脑干神经核团受损所致,随着病情的进展,临床表现逐渐明显且多见。
震颤
主要表现为运动性震颤、姿势性震颤或意向性震颤,若伴有锥体外系损害,也可出现静止性震颤。
痉挛状态
由锥体束受损所致,表现为躯干及肢体肌张力增高、腱反射活跃或亢进、踝阵挛、Babinski征阳性等,行走时呈明显的痉挛性步态。
锥体外系症状
部分患者由于基底节受损,故可伴发帕金森病样表现,或出现面、舌肌搐颤,肌阵挛、手足徐动症、扭转痉挛、舞蹈样动作等锥体外系表现。
认知功能及精神障碍
表现为注意力、记忆力受损,任务执行功能下降,其中抑郁、睡眠障碍、精神行为异常、偏执倾向是临床常见的精神障碍。
其他症状视神经病变:原发性视神经萎缩、视网膜色素变性等症状可见于常染色体显性遗传性共济失调Ⅱ型、Friedreich共济失调、共济失调⁃毛细血管扩张症、植烷酸贮积病等亚型,患者多伴有视力、视野及瞳孔改变。
骨骼畸形:为常见体征,主要表现为脊柱侧弯或后侧凸,少数患者还可发生爪形手或隐性脊柱裂等畸形,尤其是Friedreich共济失调患者,以弓形足及脊柱弯曲最常见。
皮肤病变:多见于眼球结膜、面颈部皮肤毛细血管扩张、皮肤鱼鳞症、牛奶咖啡色素斑等表现,常见于共济失调-毛细血管扩张症或Refsum综合征患者。
神经系统外的其它症状还包括心肌肥厚、糖、脂肪酸、磷脂、维生素代谢异常。
就医大部分患者因为步态不稳、吐字不清、吞咽困难和饮水呛咳、震颤、躯干及肢体肌张力增高、腱反射活跃或亢进,面、舌肌搐颤,肌阵挛、手足徐动症、扭转痉挛、舞蹈样动作就诊,通过实验室检查、影像学检查、神经系统检查等进行诊断。
就医指征出现步态不稳、吐字不清、吞咽困难和饮水呛咳、书写障碍、站立不稳,注意力、记忆力受损,抑郁、睡眠障碍、精神行为异常、偏执倾向等,尤其是有遗传性共济失调家族史患者,需要及时就医。
就诊科室该病患者应优先考虑至神经内科就诊。
医生询问病情症状是什么时候出现的?
目前都有什么症状?(如步态不稳、吐字不清、吞咽困难和饮水呛咳、书写障碍、站立不稳等)
有无遗传性共济失调家族史?
是否有既往病史?
是否去其他医院就诊过?
需要做的检查神经系统检查
进行小脑功能、肌肉力量、感觉、反射等方面的检查。
影像学检查
CT、MRI可显示是否出现小脑或脑干萎缩,患者一般出现颈髓萎缩。
实验室检查
包括血糖、脂肪酸、磷脂、维生素代谢等检查,但以下遗传性共济失调患者伴有特异性的生化指标异常。
共济失调伴肌阵挛或肌阵挛癫,包括线粒体脑肌病、蜡样脂质褐质沉积病(ceroid lipofuscinosis)、唾液酸沉积症(sialidosis)等。
肝豆状核变性(HLD),有些肝豆状核变性患者小脑体征十分显著,血清铜蓝蛋白检测有助于诊断。
β脂蛋白缺乏症,与维生素E吸收障碍有关,但随着年龄的增长其症状可逐渐减弱。光学显微镜下常可发现棘红细胞,且血清中不能检测到β脂蛋白。
脑腱黄瘤病,以年轻人好发,主要表现为痉挛-共济失调综合征、动脉粥样硬化、白内障。腱黄瘤的存在和血清高胆甾烷醇水平有助于诊断颅内可能存在的黄瘤。
神经电生理检查
可检测出患者是否出现体感诱发电位、听觉诱发电位、视觉诱发电位、眼震电图、神经肌电图等异常。
诊断标准有遗传性共济失调的家族史
医生会询问患者是否有该病家族史,帮助进行疾病诊断。
神经系统表现
包括步态不稳、吐字不清、吞咽困难和饮水呛咳、书写障碍、站立不稳等,亦可伴大脑皮质功能损害,如认知功能障碍和(或)精神行为异常等。还可以出现神经系统以外的病变,如心脏病变、内分泌代谢异常、骨骼畸形、皮肤病变等。
CT、MRI提示小脑萎缩
有时可见脑干萎缩,脑干诱发电位可出现异常,肌电图显示周围神经损害。
实验室检查
血糖、脂肪酸、磷脂、维生素代谢,部分患者可出现异常。
神经电生理检查
出现眼震电图、神经肌电图等异常,帮助该病进行诊断。
鉴别诊断遗传性痉挛性截瘫
遗传性痉挛性截瘫患者可有共济失调障碍等神经系统表现,可通过基因检测与遗传性共济失调进行鉴别。
中毒性共济失调
患者有共济失调障碍,多由于酒精、药物、重金属所致,且无家族遗传史。而遗传性共济失调有家族史,可以此进行鉴别。
治疗目前尚无能够完全阻止遗传性共济失调病情进展的治疗,遗传性共济失调的临床治疗仍以对症治疗和支持治疗为主,主要目标是减轻症状、缓解病情进展、维持日常生活自理能力。
治疗周期 患者需要长期持续性治疗。 药物治疗5-羟色胺1A受体激动药改善共济失调症状
如丁螺环酮可部分改善轻度小脑共济失调症状,坦度螺酮治疗脊髓-小脑性共济失调3型部分有效。
左旋多巴
可通过血⁃脑脊液屏障进入中枢神经系统,经多巴脱羧酶作用转化为多巴胺从而改善肌强直、运动减少等症状。苯海索对中枢神经系统胆碱受体有阻断作用,可改善肌强直、运动减少等症状。
抗癫痫药物
抗癫痫药物卡马西平可较好控制患者的癫痫发作症状。目前,对于患者所伴随的构音障碍症状尚无有效的对症治疗药物,可通过言语矫正训练进行改善。
扩张血管和改善循环药物
可扩张外周血管,改善血液循环,如烟酸、维生素E、烟酸酯、环扁桃酯、己酮可可碱等。
神经元活化药物
辅酶Q10可促进神经元代谢和呼吸功能,促进氧化磷酸化,具有抗氧化、保护生物膜结构完整性的功效。
维生素类药物
对维持神经元正常代谢过程和改善功能有一定作用。可服用B族维生素、维生素E等。
手术治疗本病无手术治疗。
心理治疗主要采取认知治疗,以改变患者非理性信念,改善认知曲解及负性思维,唤起患者的正性情感,使其发挥自身能动性。
其他治疗认知功能障碍
目前尚无有效的药物治疗,对患者早期的心理治疗策略包括认知行为干预治疗,有助于症状出现后的积极应对。
神经康复
步态不稳的患者可以接受平衡功能训练,帮助患者恢复正常生活。
预后该疾病目前不可治愈,发现疾病时应及时延缓病情进展,缓解临床症状,维持日常生活自理能力。部分患者需要终身进行康复治疗。
能否治愈该病目前不可以治愈。
能活多久该病会影响自然寿命。
复诊任何患者均需进行长期遵医嘱随访,部分患者需要终生康复治疗。
饮食 饮食调理该疾病并无特殊饮食禁忌,正常饮食即可。
护理遗传性共济失调患者需注意休息,少熬夜,保持良好的生活习惯,注意改善生活环境,家属应加强与患者交流,对患者进行相应的心理疏导,注意保持卫生。
日常护理注意保持卫生,改善生活环境。
加强与患者交流与心理指导。
病情监测日常应注意监测患者步态不稳、吐字不清、认知功能障碍等症状是否有加重,如有加重需及时到医院就医。
特殊注意事项患有该病的患者在日常生活中需特别注意,需要别人照顾,注意摔伤及跌倒。
预防遗传性共济失调预防的重点是进行遗传咨询,产前诊断或胚胎植入前诊断是目前控制遗传性共济失调发病的最佳手段。
早期筛查孕前需要进行遗传咨询。
预防措施避免近亲结婚,携带者需要进行基因检测。
产前诊断和胚胎植入前诊断是目前控制该疾病的最佳手段。
参考文献
[1]中华医学会神经病学会神经遗传学组.遗传性共济失调诊断与专家共识[J].中华神经科杂志.2015,48(6):459-463.
[2]耿德勤,刘春风.遗传性神经系统疾病-遗传性共济失调[J].中国医师进修杂志,2006(22):4-6.
本文出处:https://jk.yebaike.cn/view/176.html
 微信扫一扫
微信扫一扫