先天性胆道闭锁
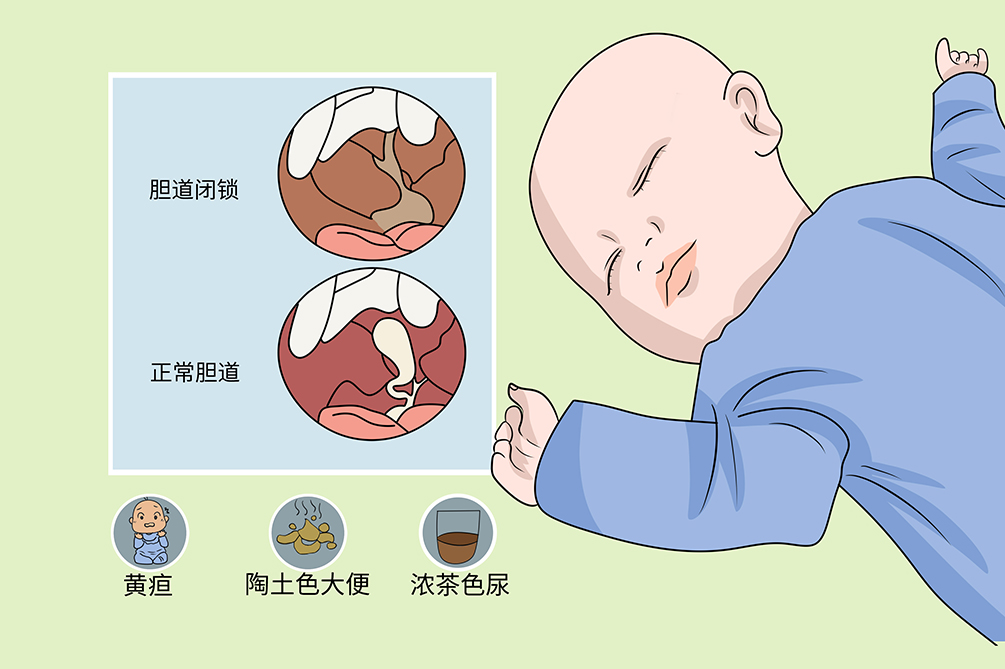
先天性胆道闭锁是新生儿一种胆管闭塞性疾病,仅见于婴儿,多与肝内外胆道先天发育异常或肝内外胆道感染致胆汁排泄障碍有关,病理性黄疸、陶土色大便、浓茶色尿是主要临床表现,目前提倡8周前早期手术,手术治疗的目的是重建、疏通或恢复胆道通畅,改善肝脏淤胆。手术治疗及时半数患儿可生存5年,不进行手术治疗,患儿一般在2年内死亡。
肝外胆道闭锁主要分为三型:
Ⅰ型
只涉及胆总管。
Ⅱ型
肝胆管闭锁。
Ⅲ型
肝门部胆管闭锁。
病因先天性胆道闭锁的病因和发病机制极为复杂,至今未完全阐明。该病是一种多病因、多表型的严重疾病,不同的类型其病因也不尽相同。先天性胆道闭锁主的发病过程中有多种触发因素,如感染、嵌合体、基因缺陷等,再加上自身免疫失调,从而导致胆道的免疫损伤扩大化。
先天发育异常
发育异常假说主要针对于伴有其他器官畸形的先天性胆道闭锁,一般认为胆道病变起于妊娠5~6周,而并非肝内胆管发育期(妊娠7~10周),胚胎细胞的分化和形态发生是器官发育的关键部分。
病毒感染
妊娠期间宫内病毒感染可导致胆管发生炎症,出现胆道纤维化,最后闭塞,常见的病毒有呼吸道病毒、巨细胞病毒、轮状病毒等。
免疫因素
患儿体内巨细胞数量增多,自身免疫细胞攻击胆道上皮细胞,导致肝内胆管变性。另外,患儿的免疫细胞也会攻击发育异常的胆管上皮细胞,最终导致胆道闭锁。
肝外胆道血供
由于肝外胆道血供出现问题,导致胆道损伤扩大化,胆道病变由纤维化、炎性病变最终进展为胆道闭锁。
母亲妊娠期解除有毒物质,会诱发新生儿先天性胆道闭锁。
婴儿免疫系统受损后容易受病毒感染,诱发新生儿先天性胆道闭锁。
性别及种族因素,亚洲女婴多见。
先天性胆道闭锁多见于早产的婴儿。
流行病学先天性胆道闭锁是一种罕见的胆管疾病,仅见于婴儿,且女婴多于男婴。该病发病呈明显的地区和种族差异,我国的发病率约为1:8000 ~1:15000。
好发人群自身基因缺陷者
患儿基因缺陷致免疫系统异常,自身免疫细胞攻击自身胆道细胞。
免疫功能低下者
妊娠期发生呼吸道病毒、巨细胞病毒、轮状病毒感染的产妇,所娩出的新生儿免疫功能低下时,病毒可由母体感染至新生儿,导致发病。
亚洲女婴
母亲妊娠期糖尿病者
症状先天性胆道闭锁患儿表现为持续的黄疸、胆汁性肝硬化,梗阻性黄疸多于生后不久出现,并呈进行性加重,同时伴有陶土色大便、浓茶色尿等症状。
黄疸
本病突出的表现是梗阻性黄疸,出生1~2周后的新生儿,本该逐步消退的生理性黄疸反而更加明显,呈进行性加重,巩膜和皮肤由金黄色变为绿褐色或暗绿色。大便渐为陶土色,尿色加深呈浓茶样,尿布染黄,皮肤有瘙痒抓痕。
营养及发育不良
初期病儿情况良好,营养发育正常,临床表现与黄疸程度不相符。随后一般情况逐渐恶化,至3~4个月时出现营养不良、贫血、发育迟缓、反应迟钝等。
肝脾大
出生时肝脏正常,随病情发展而呈进行性肿大,2~3个月即可发展为胆汁性肝硬化及门静脉高压症,发生出血倾向及凝血功能障。最终出现感染、出血、肝衰竭,严重时死亡。
其他症状部分患儿出现肝脾肿大,表现为腹部明显膨隆。
有些患儿出现消化功能变差、营养欠佳、精神萎靡。
并发症生长发育不良
由于肝脏功能受损,胆汁排放量减少,导致肠道内脂肪分解、营养物质吸收能力差,再加上肝脏本身功能不全,导致营养不良和生长发育减缓等问题。
肝功能受损
瘀积的胆汁反复刺激肝脏细胞,导致瘢痕组织代替了正常的肝组织,加之胆道损伤扩大化,胆道病变由纤维化、炎性病变最终进展为胆道闭锁,从而导致肝功能受损。
食管静脉曲张
肝硬化导致食管静脉曲张,可并发破裂、出血。
就医婴儿出现持续的黄疸不消退,并出现巩膜和皮肤呈绿褐色或暗绿色时,应及时就医。通过相关检查,如血液检查、B超检查等可以明确诊断。
就医指征生理性黄疸消退后又出现眼白、皮肤发黄,呈进行性加重,随着黄疸加重,大便颜色呈陶土样,尿色呈浓茶色应及时就医。
患儿3月龄后生长发育减缓,消化功能变差,营养不良,精神差,5~6月龄出现皮肤干燥、皮下淤血、鼻出血等情况应及时就医。
大多患者优先考虑去儿科就诊。
若患者出现其他严重不适反应或并发症,如精神萎靡、生长发育不良等,可到相应科室就诊,如神经科或外科等。
医生询问病情黄疸是一过性还是持续的?
目前都有什么症状?(如精神萎靡,喂养困难等)
婴儿尿液颜色是否正常?
出现这些症状有多久了?
患儿最近是否出现烦躁不安、易怒等?
需要做的检查粪便比色卡筛查
粪便比色卡筛查是全面评估黄疸患儿的一个关键环节,可以使胆道闭锁Kasai手术日龄提前。
血液检查
虽然单纯的肝功能检查对诊断胆道闭锁缺乏特异性,但可以反映肝脏的损害程度,且这些检查为无创血液检查,对于诊断先天性胆道闭锁有一定的临床意义。
体格检查
检查患儿皮肤和眼白,判断是否出现黄疸;对患儿进行腹部触诊,判断患儿肝脾是否肿大;检查患儿是否伴有其他出生缺陷。
B超
超声检查可用于胆道闭锁早期筛查,胆囊形态不规则、囊壁僵硬而毛糙、厚度不均,收缩功能改变可做为筛查指标。B超因无创而成为肝胆疾病的首选检查项目,可显示胆囊是萎陷还是缺如。
腹腔镜探查辅助胆道造影
腹腔镜可直接观察腹腔中肝脏和胆管的具体情况,胆道造影可确诊胆道闭锁。
肝活检
肝活检属于有创检查,且肝纤维化分布不均匀可造成取样误差而影响肝活检的准确性,但是肝活检有助于排除其他肝胆疾病。
诊断标准出生后1-2个月出现持续性黄疸,陶土色大便、深茶色尿,伴肝大者均应怀疑本病,以下有助于确诊:
黄疸超过3~4周仍呈进行性加重,对利胆药物治疗无效;对苯巴比妥和激素治疗无反应;以直接胆红素升高为主的血清胆红素动态观测呈持续上升。
十二指肠引流液内无胆汁。
超声检查显示肝外胆管和胆囊发育不良或缺如。
99mTe-EHIDA扫描肠内无核素显示。
ERCP和MRCP显示胆管闭锁。
鉴别诊断婴儿胆汁淤积症
又称为婴儿肝炎综合征,也表现为黄疸、大便颜色变浅等,可通过超声等影像学检查或组织病理检查进行鉴别。
生理性黄疸
大部分足月儿出生后3-5天会出现黄疸,但不严重,且于2周内自行恢复,早产儿生理性黄疸于3周以上自行恢复,但胆道闭锁引起的黄疸多出现在生理性黄疸消退后,且呈进行性加重,故可鉴别。
治疗先天性胆道闭锁患儿主要通过手术治疗辅以药物治疗,肝门空肠吻合术为首选手术治疗方案,术后辅助肝移植手术可提高患儿存活率。
补充脂溶性维生素A、D、E、K,因胆汁的肠肝循环障碍而导致脂溶性维生素吸收障碍,故需要补充。
给予退黄、保护肝功能、免疫球蛋白调节免疫等对症治疗。
手术治疗的患儿给予患者营养支持,由于患儿在手术前和手术后需要禁食,难免导致患儿身体虚弱以及营养不良。故而,在患儿手术之后,应该对其进行必要的营养支持,以维持机体正常的生长发育,促进患者伤口的愈合。
药物治疗雄去氧胆酸
口服熊去氧胆酸可增加胆汁酸分泌,降低胆汁中胆固醇及胆固醇酯,有利于肝脏排出胆汁。
激素
激素可以抑制毛细血管扩张,减轻渗出和水肿,又可抑制白细胞的浸润和吞噬,而减轻炎症反应,从而促进胆汁排出。
抗生素
先天性胆道闭锁患儿的抵抗力较低,极易导致临床感染的发生。故而,在患儿手术之后,应该积极的预防其发生感染,加强消毒与隔离,并给予其必要的抗生素等。
手术治疗肝门空肠吻合手术
先检查胆囊并穿刺了解胆汁性状,游离胆囊,结扎胆囊动脉,然后在肝被膜表面彻底剪除肝门纤维块。距离treitz韧带15cm处断离空肠,然后将肝脏直接和空肠吻合在一起,使肝脏产生的胆汁顺利排入肠道。患儿最好于2个月前行肝门空肠吻合手术。
肝移植
切除患儿的肝脏,用健康的肝脏替代。适用于肝门空肠吻合手术失败、术后并发症经久不愈的患儿。术后应服用免疫抑制药物,避免发生免疫排斥反应,术后多数患儿可延长5~10年寿命。
预后先天性胆道闭锁患儿如果不进行手术治疗,一般会在2年内死亡;接受肝门空肠吻合手术的患儿,半数存活率为5年,但伴有严重的肝脏并发症的患儿,生存期会明显缩短,行肝门空肠吻合手术加肝移植的患儿,预后较好,可有5~10年的存活率。
先天性胆道闭锁为终身疾病,尚不能治愈。
能活多久先天性胆道闭锁患儿未进行手术治疗,寿命不超过2年;行肝门空肠吻合手术,寿命不超过5年;行肝门空肠吻合手术加肝移植手术,寿命不超过10年;若期间合并严重的并发症,寿命会明显缩短。
复诊每日注意观察患儿粪便颜色、精神状况等,如出现不明原因的发热、出血,应立即就诊;每日测量、关注患儿腹围;术后每半月至一月行血液检查体内蛋白指标。
饮食先天性胆道闭锁患儿的饮食需保证患儿生长发育所需的足够的营养和热量,患儿还需补充脂溶性维生素和中链甘油三酸酯。患儿消化差,容易饿,喂养需要少食多餐。
饮食调理术后宜母乳喂养,如母乳不充足,应用短链脂肪酸类奶粉进行喂养。
护理先天性胆道闭锁患儿术前以及手术后的临床护理工作是保障临床治疗效果的重要手段,对患儿进行积极的护理,可有效减少患儿并发症的发生,提高了患儿的生活质量。
日常护理避免到人多的公共场合,避免接触患病的人而受到感染,避免受凉等。
每日关注、测量患儿腹围、患儿皮肤及黏膜颜色、患儿粪便颜色等。
家长应戒烟,减少二手烟对患儿的伤害。
病情监测患儿家长应每天关注患儿皮肤肤色情况,是否存在复发或加重等。
养成观察患儿大小便的习惯,注意观察大小便颜色情况,如有异常及时就医。
患儿由于长期服用免疫抑制剂,故家长应注意患儿有无感染的情况。
特殊注意事项患儿生后2~3周,如果黄疸复现且呈进行性加重,家长应及时就医排除胆道闭锁的情况。
对于肝门空肠吻合术后患儿,若出现黄疸复现、大便颜色变浅、发热、腹痛等症状,需警惕胆管炎的发生,此术后并发症常威胁生命。
肝移植术后患儿复现黄疸进行性加重,粪便颜色变浅、发热、腹痛等,应警惕出现免疫排斥反应,需及时就医。
预防由于先天性胆道闭锁的确切病因尚未明确,妊娠期间避免感染和有毒物质接触,及出生后做好新生儿早期筛查等,可在一定程度上避免先天性胆管闭锁的发生。
早期筛查妊娠期发生呼吸道病毒、巨细胞病毒等感染的孕妇,分娩后应早开始进行新生儿阻塞性黄疸的筛查。
妊娠期间保证充足睡眠,增强自身免疫力,避免病毒感染。
妊娠期注意避免有毒气体及物质的接触。
新生儿应避免去人多且环境复杂的场所,以免发生感染。
参考文献
[1]范艳华,韩春霞.68例先天性胆道闭锁患儿的术后效果及影响因素分析[J].内蒙古医学杂志,2019.
[2]郭振亚.先天性胆道闭锁的病因研究进展[J].临床肝胆病杂志,2015.
[3]伍新平.先天性胆道闭锁患儿的手术护理配合[J].全科护理,2018.
[4]胡萍.小儿先天性胆道闭锁33例Kasai手术治疗体会[J].陕西医学杂志,2012.
本文出处:https://jk.yebaike.cn/view/680.html
 微信扫一扫
微信扫一扫